
¿Qué son?
Las enfermedades neuromusculares son enfermedades
neurológicas, de naturaleza progresiva, normalmente hereditarias y su principal
característica clínica es la debilidad muscular.
Son un conjunto de más de 150 enfermedades neurológicas, de
naturaleza progresiva, en su mayoría de origen genético y su principal
característica es la pérdida de fuerza muscular. Son enfermedades crónicas que
generan gran discapacidad, pérdida de la autonomía personal y cargas psicosociales.
Todavía no disponen de tratamientos efectivos, ni curación. Su aparición puede
producirse en cualquier etapa de la vida, pero más del 50% aparecen en la
infancia. En cifras globales, existen más de 60.000 afectados por enfermedad
neuromuscular en toda España.
El músculo, la unión neuromuscular (donde se junta el nervio
con el músculo), el nervio periférico (en brazos, piernas, cuello y cara), La
motoneurona espinal (células nerviosas que controlan la acción de los
músculos). Su aparcición puede producirse en cualquier etapa de la vida, tanto
en el nacimiento como en la adolescencia o en la edad adulta.
SE ENCUENTRAN DENTRO DEL GRUPO DE LAS DENOMINADAS
ENFERMEDADES RARAS.
Tipos de enfermedades neuromusculares
No existe un único criterio a la hora de clasificar las
Enfermedades Neuromusculares ya que puede hacerse desde el punto de vista
fisiopatológico, clínico o bien dar prioridad a otros conceptos como la forma
de transmisión hereditaria. Actualmente tiene interés la clasificación basada
en la biología molecular, lo que permite la creación de nuevos subtipos dentro
de un mismo conjunto de síntomas.
Los principales tipos de enfermedades neuromusculares son:
1. Distrofias musculares
2. Miopatías distales
3. Miopatías congénitas
4. Distrofia miotónica de Steinert
5. Miotonías congénitas
6. Parálisis peródicas familiares
7. Enfermedades musculares inflamatorias
8. Miositis osificante progresiva
9. Miopatías metabólicas
10. Enfermedades de la unión neuromuscular
11. Amiotrofias espinales
12. Neuropatías hereditarias sensitivo-motoras (enfermedades de Charcot-Marie-tooth)
2. Miopatías distales
3. Miopatías congénitas
4. Distrofia miotónica de Steinert
5. Miotonías congénitas
6. Parálisis peródicas familiares
7. Enfermedades musculares inflamatorias
8. Miositis osificante progresiva
9. Miopatías metabólicas
10. Enfermedades de la unión neuromuscular
11. Amiotrofias espinales
12. Neuropatías hereditarias sensitivo-motoras (enfermedades de Charcot-Marie-tooth)
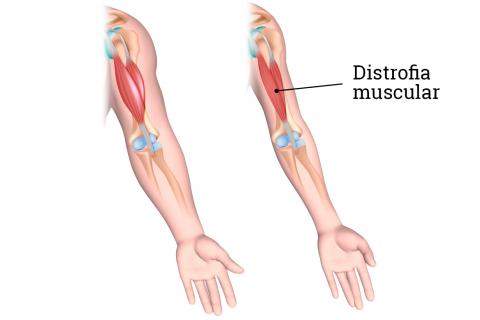
Distrofias musculares
Las distrofias musculares (DM) afectan predominantemente al
músculo estriado y son debidas a un defecto alguna de las proteínas que forman
parte de la fibra muscular, ya sean estructurales o enzimáticas (ejemplos son
la distrofina calpaína, merosina y emerina, entre otras).
Dentro de este grupo se distinguen varias categorías:
Distrofinopatías
Son distrofias musculares progresivas que se caracterizan
por anomalías moleculares de la distrofina, una de las principales proteínas
que mantienen la estructura de la fibra muscular. Tienen una forma de
trasmisión ligada al cromosoma X, por lo que la trasmiten las mujeres y la
manifiestan clínicamente los hombres.
Dentro de este subgrupo existen varias formas clínicas:
Distrofia Muscular de Duchenne:
Se caracteriza por una debilidad progresiva de la cintura
pélvica en la infancia (a partir de los 2 ó 3 años). Las características de la
postura que adopta el niño son: Torso hacia atrás, marcha dandinante,
dificultad para subir las escaleras.
Suele haber una pérdida de la marcha entre los 10 y los 13
años y una agravación y generalización de la afectación muscular, incluidos y
el músculo cardíaco. A partir de la adolescencia, suele requerir asistencia
respiratoria.

Distrofia muscular de Becker:
Los síntomas son muy parecidos a los de la DM de Duchenne,
aunque su intensidad es menor y su aparición es más tardía.
La progresión es más lenta y la esperanza de vida es más
prolongada que el fenotipo Duchenne.
Distrofias musculares congénitas
Son distrofias musculares que por alteración de proteínas
musculares que se manifiestan clínicamente desde el nacimiento o en los
primeros meses de vida. Cursan con hipotonía, debilidad de los músculos de los
miembros y del tronco, y pueden asociar otro tipo manifestaciones como
retracciones musculares, malformaciones oculares o alteraciones de la sustancia
blanca cerebral. El modo de herencia es variable.
Distrofia muscular de Emery-Dreifuss
Es una distrofia muscular progresiva con patrón de herencia
autosómico dominante que suele manifestarse entre la 1º y 2º década de la vida.
Se caracteriza por la aparición de retracciones del bíceps, del tendón de
Aquiles y de los músculos cervicales posteriores. Cursa con debilidad muscular
húmero-peroneal y afectación cardíaca: arritmias, problemas de conducción o
insuficiencia de la función ventricular, que pueden ser letales si no se
detectan y se tratan
Su progresión es lenta con aparición de debilidad muscular
en hombros y brazos.
Distrofias musculares de cinturas (LGMD)
Se trata del grupo más heterogéneo desde el punto de vista
clínico como molecular. Inicialmente se agruparon bajo este nombre aquellas
distrofias musculares con debilidad fundamental de la cintura pelviana o
escapular, y que no correspondían al fenotipo Duchenne o facioescapulohumeral.
Se dividen en dos grandes grupos según su modo de herencia autosómico dominante
(LGMD1) o recesivo (LGMD2).Las formas más graves suelen corresponder al grupo
de las recesivas, como las debidas al déficit de sarcoglicanos, calpaína o
disferlina. En el grupo de las dominantes existen formas más leves, incluyendo
hiperCKemia asintomática (como sucede en algunas formas por déficit de
caveolina-3).
Distrofia muscular facioescápulo humeral
Es una de las distrofias musculares más frecuentes, y tiene
un patrón de herencia autosómico dominante. Suele manifestarse en la juventud,
aunque existen formas de inicio más tardío y la penetrancia es variable. Se
caracteriza por la presencia de debilidad y atrofia de los músculos de la cara
y de la cintura escapular: movilidad facial reducida, dificultad para levantar
los brazos por encima de la cabeza, hombros caídos hacia delante y omóplatos
prominentes.
Los glúteos y músculos anteriores de la pierna están
afectados.
Su evolución es muy lenta con frecuentes períodos de
estabilización.
La esperanza de vida es normal a pesar de que la incapacidad
funcional es, a menudo, grave.
Miopatías distales Subir
Son un grupo de enfermedades con patrón de herencia
autosómico recesivo o dominante. La afectación es predominantemente en la
musculatura distal de miembros inferiores, y según el tipo será afectará
fundamentalmente al compartimento anterior o posterior de las piernas. Algunas
de ellas se caracterizan por presenta vacuolas ribeteadas en la biopsia
muscular.
Son progresivas (los síntomas se gravan con el paso del
tiempo) con una afectación de los músculos ascendente, pero el ritmo de
evolución es moderado.
Dentro de este grupo se encuentran
- Miopatía distal de tipo Welander
- Miopatía distal de tipo Markesbery-Griggs
- Miopatía distal de tipo Miyoshi
- Miopatía distal de tipo Nonaka
Miopatías congénitas Subir
Dentro de este grupo se distinguen varios tipos de
enfermedades, con patrón de herencia variable. Estas enfermedades se producen
por un defecto en el desarrollo del músculo, lo cual produce unas alteraciones
características en la biopsia muscular, específicas de cada uno de los tipos de
miopatía congénita. Suelen diagnosticarse poco después del nacimiento (de ahí
lo de congénitas) al observar que el bebé se mueve poco, está débil y adopta
posiciones anormales o no se alimenta correctamente.
Algunos tipos pueden aparecen en la infancia, la
adolescencia o la edad adulta.
Dentro de este grupo se encuentran:
Miopatía congénita nemalínica (Nemaline myopathies)
Se caracteriza por la aparición de una hipotonía
generalizada y difusa con afectación de manos, pies, tronco y cara.
En el lactante, existen variantes en las que los músculos
respiratorios se ven afectados.
El déficit no es evolutivo y, en general, causa invalidez
moderada, en particular en los niños mayores y en los adultos.
En ocasiones aparece un déficit importante con deformaciones
ortopédicas, insuficiencia respiratoria y problemas de deglución.
Miopatía congénita “central core”
Se caracteriza por la presencia de debilidad muscular
predominante en los hombros y en la pelvis. El inicio de la deambulación suele
ser tardío.
En ocasiones existe dificultad para correr, para subir
escaleras y escoliosis (desviación de la columna vertebral).
La afectación no es progresiva produciendo una invalidez
moderada.
Miopatía congénita centronuclear
Se caracteriza por la presencia de debilidad muscular de las
piernas y de la cara.
Suele haber un retraso en el comienzo de la marcha.
Su evolución es variable y la enfermedad puede causar
distintos grados de invalidez.
Miopatía congénita miotubular
Cursa con una hipotonía en el recién nacido e insuficiencia
respiratoria importante.
El afectado suele fallecer en las primeras semanas de vida.
Miopatía congénita con minicores
Se caracteriza por una hipotonía neonatal e inmovilidad de
la cara. En ocasiones existe deformación del tórax y de los pies, e incluso a
veces afección cardíaca.
Su evolución es muy variable, más o menos lenta.
Distrofia miotónica de Steinert Subir
Es la distrofia muscular más frecuente. Tiene una herencia
autosómica dominante, y se produce el fenómeno de anticipación: los síntomas
suelen aparecer de forma más precoz y suelen ser más graves en generaciones
sucesivas. Existen formas congénitas (muy graves y a menudo letales) y formas
de inicio más tardío.
Se caracteriza por la aparición de una debilidad progresiva
de los músculos faciales, elevadores de párpados (existe ptosis), bulbares
(suele existir disfagia) distales de extremidades rigidez miotónica. Lo que
caracteriza y da nombre a esta enfermedad es la dificultad para relajar los
músculos después de una contracción mantenida, lo que se denomina “fenómeno
miotónico”.
Es habitual la presencia de cataratas, calvicie y anomalías
endocrinas, hormonales y cardíacas (imprescindible hacer un seguimiento
cardiaco periódico).
Su evolución es variable y puede llegar a alcanzar un estado
de gran invalidez a los 15 o 20 años tras su aparición.
Miotonías congénitas
Se manifiestan desde el nacimiento o en la infancia.
Dentro de este grupo se encuentran:
Miotonías congénitas
Autosómica recesiva (o tipo
Becker, más grave) y autosómica dominante (o tipo Thomsen, más leve).
Es una miotonía difusa que se agrava con el frío y mejora
con el movimiento (después de una contracción, aparece una lentitud anormal de
la relajación muscular referida como contractura muscular).
Miotonía condrodistrófica (síndrome de Schwartz– Jampel)
Es una miotonía, en ocasiones dolorosa, asociada a problemas
del crecimiento. Por eso, son frecuentes las deformaciones esqueléticas.
Parálisis periódicas familiares subir
Dentro de este grupo se encuentran:
Adinamia episódica de Gamstorp y enfermedad de Westphal
Se manifiesta con episodios de parálisis, con una duración y
frecuencia variables. Afectan a los cuatro miembros, y son provocados por: el
descanso tras el ejercicio, una comida muy salada y/o rica en azúcares, la
exposición al frío, un episodio febril o un traumatismo físico o psíquico. En
general, no existen molestias entre las crisis.
Se observa una mejoría con la edad: desaparición de las
crisis hacia los 40 – 50 años.
Paramiotonía de Eulenburg
Miotonía persistente con el ejercicio, que se agrava
visiblemente con el frío y va acompañada de debilidad muscular. Es una
afectación estable.
Enfermedades musculares inflamatorias Suibir
Se trata de un grupo de enfermedades adquiridas (no
hereditarias) de causa inmunológica.
Dentro de este grupo encontramos:
Polimiositis y Dermatomiositis
Son enfermedades inflamatorias del músculo que aparecen en
la infancia o en la edad adulta. Se caracterizan por la presencia de mialgias y
debilidad de los músculos predominantemente proximales (hombros, pelvis y
cuello).Suele aparecer una erupción eritematosa en la cara y en la parte alta
del tronco (en la dermatomiositis).Su evolución es variable, aunque a menudo
suele ser rápida, y grave si no se tratan. Lo más importante es que se trata de
enfermedades tratables, y suelen responder bien a inmunosupresores.
Miositis por cuerpos de inclusión
Es una enfermedad inflamatoria del músculo con un comienzo
insidioso en la edad adulta. Se caracteriza por la presencia de debilidad
muscular y amiotrofia proximal de los miembros inferiores, en general
simétricos, de musculatura flexora de la mano y de músculos bulbares (es
frecuente la disfagia) A pesar de ser una enfermedad de supuesta etiología
inmune no responde bien a ninguno de los tratamientos ensayados.
Miositis Osificante progresiva Suibir
Es una enfermedad que se manifiesta en la infancia. Se
producen crisis de osificación de los músculos que se vuelven “duros como
piedras” (el hueso empieza a comer al músculo). Estas osificaciones producen
limitaciones articulares y deformidades.
Su evolución se produce por brotes o por crisis a lo largo
de toda la vida.
Miopatías metabólicas Suibir
Se trata de un grupo de enfermedades genéticamente
determinadas, cuya base etiopatogénica es la dificultad para obtener energía
por parte de la fibra muscular.
Este grupo se clasifica, a su vez, en 3 subgrupos:
Miopatías mitocondriales
Son miopatías metabólicas que aparecen en la primera
infancia o en la edad adulta (según el tipo).
En la mayoría de los casos, aparece debilidad permanente de
los músculos de los ojos (caída palpebral o ptosis) con o sin afección muscular
de los miembros. También es frecuente la presencia de fatigabilidad, a menudo
dolorosa con el esfuerzo. Puede aparecer, de forma progresiva, oftalmoplejía.
Los afectados suelen tener dificultad para alimentarse y
problemas de deglución (en las formas graves). Su evolución es variable según
la gravedad del tipo, aunque la mayoría no causan demasiada invalidez.
En las formas graves, es posible una afección cerebral que
pueda causar problemas de equilibrio, epilepsia y/o parálisis.
Lipidosis musculares
Son miopatías metabólicas que aparecen en el recién nacido,
en la infancia o en la edad adulta, según la forma.
Glucogenosis musculares
Se trata de miopatías metabólicas que pueden aparecer a
cualquier edad: desde la infancia a la edad adulta.
Suelen cursar con fatiga muscular, dolores al realizar
esfuerzo y calambres.
Enfermedades de la unión neuromuscular Suibir
Dentro de este grupo se engloban tres subgrupos:
Miastenia Gravis
Se trata de una enfermedad de causa inmunológica, producida
por la presencia de anticuerpos contra componentes de la membrana postsináptica
de la unión neuromuscular. Es una enfermedad que puede manifestarse a cualquier
edad, aunque lo más frecuente es entre los 20 y 30 años en las mujeres y entre
los 40 y 60 en los hombres.
Se caracteriza por presentar una debilidad muscular de
intensidad y duración variables que pueden afectar a cualquier músculo. Esta
debilidad puede aumentar con el esfuerzo y/o con la repetición del movimiento.
Su evolución es variable, con remisiones o exacerbaciones.
Se trata con inmunosupresores.
Síndrome de Eaton-Lambert
Es una enfermedad debida a anticuerpos contra componentes de
la membrana presináptica de la unión neuromuscular. En un porcentaje elevado de
las ocasiones se trata de una enfermedad paraneoplásica, por lo que se debe
descartar la presencia de un cáncer oculto (especialmente de pulmón). Produce
debilidad en MMII, y alteraciones autonómicas (impotencia, estreñimiento…)
Síndromes miasténicos congénitos
Se trata de enfermedades genéticamente determinadas, que
aparecen desde el nacimiento. Se caracterizan por la presencia de fatiga
anormal debida a una debilidad muscular localizada o generalizada.
Algunas formas responden de forma parcial a tratamiento con
anticolinestrásicos.
Existe una forma adulta de posible comienzo tardío (el
síndrome del canal lento).
Amiotrofias espinales Suibir
Las Amiotrofias espinales constituyen un grupo de
enfermedades caracterizadas por la pérdida o degeneración de las neuronas del
asta anterior de la médula espinal. El mal funcionamiento de estas neuronas
hace que el impulso nervioso no pueda transmitirse correctamente y, por tanto,
los movimientos y el tono muscular se ven afectados. Inicialmente, están mas
afectados los músculos proximales y la debilidad en los miembros inferiores
suele ser generalmente mayor que la de los miembros superiores.
Existen diferentes tipos de Amiotrofias espinales, todas con
patrones de herencia autosómico recesivo:
Amiotrofias espinales infantiles tipos I
Su inicio es antes de los 6 meses de edad y son niños con
una debilidad tan intensa que no llegan a sentarse. Se caracteriza por
presentar una debilidad simétrica de los músculos proximales y del tronco,
extendiéndose hacia las extremidades.
Aparece una parálisis de los músculos intercostales. Son
frecuentes las afecciones respiratorias.
Puede existir afectación del tronco cerebral (con un inicio
antes de los 3 meses) con riesgo de muerte súbita.
Su evolución es grave a pesar del tratamiento especializado.
Amiotrofia espinal infantil tipo II
Su inicio se produce después de los 6 meses de edad, y son
niños que llegan a poder sentarse, aunque no adquieren la marcha. Cursa con una
debilidad simétrica de los músculos proximales y del tronco. Puede haber una
posible afectación de los músculos intercostales inferiores. Su evolución es estable
tras una fase de agravación.
Amiotrofia espinal infantil tipo III
Su inicio se produce hacia el final de la infancia o
principio de la adolescencia.
Cursa con debilidad de los músculos proximales, y aunque
llegan a adquirir la marcha lo hacen con dificultad. Suele haber dificultad
para levantarse del suelo y para subir escaleras. Su evolución es variable,
habitualmente lenta.
Amiotrofia espinal del adulto tipo IV
Su inicio se produce en la edad adulta. Cursa con parálisis
y atrofia de los músculos distales de las piernas. Pueden existir dificultades
respiratorias y debilidad de los músculos de los muslos y de los antebrazos.
Neuropatías hereditarias sensitivo-motoras (enfermedades de Charcot-Marie-Tooth) Suibir
Las neuropatías hereditarias forman un grupo muy frecuente
de enfermedades genéticas. Las formas más conocidas son aquellas que se heredan
de forma autosómica dominante, que afectan a la mielina de los nervios y se
manifiestan en la infancia (enfermedad de Charcot-Marie-Tooth tipo 1A) Esta
forma se caracteriza por presentar amiotrofia distal de las piernas y los
brazos. Es frecuente una debilidad muscular que produce problemas en la marcha.
Suelen aparecer problemas de la sensibilidad profunda y superficial, dolores y,
frecuentemente, pie cavo. La gravedad de la afectación es muy variable.
Su evolución es lentamente progresiva. La limitación
funcional es muy variable de un individuo a otro.
No hay comentarios:
Publicar un comentario